To the 50th anniversary of the discovery of the structure of DNA
A.V. Zelenin
PLANT GENOME
A. V. Zelenin
Zelenin Alexander Vladimirovich- Doctor of Biological Sciences,
Head of Laboratory, Institute of Molecular Biology named after. V.A. Engelhardt RAS.
The impressive achievements of the Human Genome program, as well as the success of work on deciphering the so-called ultra-small (viruses), small (bacteria, yeast) and medium-sized (roundworm, Drosophila) genomes, made it possible to move to a large-scale study of large and extra-large plant genomes. The urgent need for a detailed study of the genomes of the most economically important plants was emphasized at a meeting on plant genomics held in 1997 in the USA [,]. Over the years since then, undoubted successes have been achieved in this area. In 2000, a publication appeared on the complete sequencing (establishment of the linear nucleotide sequence of all nuclear DNA) of the genome of the small mustard - Arabidopsis, and in 2001 - on the preliminary (draft) sequencing of the rice genome. Work on sequencing large and ultra-large plant genomes (corn, rye, wheat) has been repeatedly reported, but these messages did not contain specific information and were rather declarations of intent.
It is expected that deciphering plant genomes will open up broad prospects for science and practice. First of all, the identification of new genes and the chain of their genetic regulation will significantly increase plant productivity through the use of biotechnological approaches. The discovery, isolation, reproduction (cloning) and sequencing of genes responsible for such important functions of the plant organism as reproduction and productivity, processes of variability, resistance to adverse environmental factors, as well as homologous pairing of chromosomes, is associated with the emergence of new opportunities for improving the selection process . Finally, isolated and cloned genes can be used to obtain transgenic plants with fundamentally new properties and analyze the mechanisms of regulation of gene activity.
The importance of studying plant genomes is also emphasized by the fact that so far the number of localized, cloned and sequenced plant genes is small and, according to various estimates, varies between 800 and 1200. This is 10-15 times less than, for example, in humans.
The United States remains the undoubted leader in the large-scale study of plant genomes, although intensive research on the rice genome is being carried out in Japan, and in recent years in China. In addition to US laboratories, European research groups took an active part in deciphering the Arabidopsis genome. The apparent leadership of the United States is causing serious concern among European scientists, which they clearly expressed at a meeting meaningfully titled “Prospects for Genomics in the Postgenomic Era,” held in France at the end of 2000. The advance of American science in studying the genomes of agricultural plants and creating transgenic plant forms, according to European scientists, threatens that in the not too distant future (from two to five decades), when population growth will put humanity in the face of a general food crisis, the European economy and science will become dependent on American technology. In this regard, it was announced the creation of a Franco-German scientific program for the study of plant genomes (Plantgene) and the investment of significant funds in it.
Obviously, the problems of plant genomics should attract the close attention of Russian scientists and science organizers, as well as governing bodies, since we are talking not only about scientific prestige, but also about the national security of the country. In one or two decades, food will become the most important strategic resource.
DIFFICULTIES IN STUDYING PLANT GENOMES
Studying plant genomes is a much more complex task than studying the genome of humans and other animals. This is due to the following circumstances:
huge genome sizes, reaching tens and even hundreds of billions of nucleotide pairs (bp) for individual plant species: the genomes of the main economically important plants (except rice, flax and cotton) are either close in size to the human genome or exceed it many times (table);CHROMOSOMAL GENOME STUDIESSharp fluctuations in the number of chromosomes in different plants - from two in some species to several hundred in others, and it is not possible to identify a strict correlation between the genome size and the number of chromosomes;
An abundance of polyploid (containing more than two genomes per cell) forms with similar but not identical genomes (allopolyploidy);
The extreme enrichment of plant genomes (up to 99%) with “insignificant” (non-coding, that is, not containing genes) DNA, which greatly complicates the joining (arrangement in the correct order) of sequenced fragments into a common large-sized DNA region (contig);
Incomplete (compared to the genomes of Drosophila, human and mouse) morphological, genetic and physical mapping of chromosomes;
The practical impossibility of isolating individual chromosomes in a pure form using methods usually used for this purpose for human and animal chromosomes (flow sorting and the use of cell hybrids);
The difficulty of chromosomal mapping (determining the location on the chromosome) of individual genes using hybridization in situ, due to both the high content of “insignificant” DNA in plant genomes and the peculiarities of the structural organization of plant chromosomes;
The evolutionary distance of plants from animals, which seriously complicates the use of information obtained from sequencing the genome of humans and other animals to study plant genomes;
The long process of reproduction of most plants, which significantly slows down their genetic analysis.
Chromosomal (cytogenetic) studies of genomes in general and plants in particular have a long history. The term “genome” was proposed to denote a haploid (single) set of chromosomes with the genes they contain in the first quarter of the 20th century, that is, long before the role of DNA as a carrier of genetic information was established.
Description of the genome of a new, previously unstudied genetically multicellular organism usually begins with the study and description of the complete set of its chromosomes (karyotype). This, of course, also applies to plants, a huge number of which have not even begun to be studied.
Already at the dawn of chromosomal studies, genomes of related plant species were compared based on the analysis of meiotic conjugation (unification of homologous chromosomes) in interspecific hybrids. Over the past 100 years, the capabilities of chromosomal analysis have expanded dramatically. Nowadays, more advanced technologies are used to characterize plant genomes: various variants of the so-called differential staining, which makes it possible to identify individual chromosomes based on morphological characteristics; hybridization in situ, making it possible to localize specific genes on chromosomes; biochemical studies of cellular proteins (electrophoresis and immunochemistry) and, finally, a set of methods based on the analysis of chromosomal DNA up to its sequencing.

Rice. 1. Karyotypes of cereals: a - rye (14 chromosomes), b - durum wheat (28 chromosomes), c - soft wheat (42 chromosomes), d - barley (14 chromosomes)The karyotypes of cereals, primarily wheat and rye, have been studied for many years. It is interesting that in different species of these plants the number of chromosomes is different, but always a multiple of seven. Individual cereal species can be reliably identified by their karyotype. For example, the rye genome consists of seven pairs of large chromosomes with intensely colored heterochromatic blocks at their ends, often called segments or bands (Fig. 1a). Wheat genomes already have 14 and 21 pairs of chromosomes (Fig. 1, b, c), and the distribution of heterochromatic blocks in them is not the same as in rye chromosomes. The individual genomes of wheat, designated A, B and D, also differ from each other. An increase in the number of chromosomes from 14 to 21 leads to a sharp change in the properties of wheat, which is reflected in their names: durum, or macaroni, wheat and soft, or bread, wheat . The D gene, which contains genes for gluten proteins, is responsible for the acquisition of high baking properties by soft wheat, which gives the dough the so-called germination. It is this genome that is given special attention in the selection improvement of bread wheat. Another 14-chromosome cereal, barley (Fig. 1, d), is not usually used to make bread, but it serves as the main raw material for the production of such common products as beer and whiskey.
The chromosomes of some wild plants used to improve the quality of the most important agricultural species, for example the wild relatives of wheat - Aegilops, are being intensively studied. New plant forms are created through crossing (Fig. 2) and selection. In recent years, significant improvements in research methods have made it possible to begin studying the genomes of plants whose karyotype features (mainly small chromosome sizes) made them previously inaccessible for chromosomal analysis. Thus, only recently were all chromosomes of cotton, chamomile and flax identified for the first time.

Rice. 2. Karyotypes of wheat and wheat-Aegilops hybrid
a - hexaploid common wheat ( Triticum astivum), consisting of A, B and O genomes; b - tetraploid wheat ( Triticum timopheevi), consisting of A and G genomes. contains genes for resistance to most wheat diseases; c - hybrids Triticum astivum X Triticum timopheevi, resistant to powdery mildew and rust, the replacement of part of the chromosomes is clearly visiblePRIMARY STRUCTURE OF DNA
As molecular genetics developed, the very concept of a genome expanded. Now this term is interpreted both in the classical chromosomal and in the modern molecular sense: the entire genetic material of an individual virus, cell and organism. Naturally, after studying the complete primary structure of genomes (as the complete linear sequence of nucleic acid bases is often called) of a number of microorganisms and humans, the question of sequencing plant genomes came up.
Of the many plant organisms, two were chosen for study - Arabidopsis, representing the class of dicotyledons (genome size 125 million bp), and rice from the class of monocotyledons (420-470 million bp). These genomes are small compared to other plant genomes and contain relatively few repeated sections of DNA. Such features gave hope that the selected genomes would be accessible for relatively rapid determination of their primary structure.

Rice. 3. Arabidopsis - small mustard - a small plant from the cruciferous family ( Brassicaceae). In a space equal in area to one page of our magazine, up to a thousand individual Arabidopsis organisms can be grownThe basis for choosing Arabidopsis was not only the small size of its genome, but also the small size of the organism, which makes it easy to grow in laboratory conditions (Fig. 3). We took into account its short reproductive cycle, thanks to which it is possible to quickly conduct crossing and selection experiments, detailed genetics, ease of manipulation with changing growing conditions (changing the salt composition of the soil, adding different nutrients, etc.) and testing the effect on plants of various mutagenic factors and pathogens (viruses, bacteria, fungi). Arabidopsis has no economic value, therefore its genome, along with the mouse genome, was called a reference genome, or, less accurately, a model genome.*
* The appearance of the term “model genome” in Russian literature is the result of an inaccurate translation of the English phrase model genome. The word "model" means not only the adjective "model", but also the noun "sample", "standard", "model". It would be more correct to talk about a sample genome, or a reference genome.Intensive work on sequencing the Arabidopsis genome began in 1996 by an international consortium that included scientific institutions and research groups from the USA, Japan, Belgium, Italy, Great Britain and Germany. In December 2000, extensive information became available summarizing the determination of the primary structure of the Arabidopsis genome. For sequencing, we used classical, or hierarchical, technology: first, individual small sections of the genome were studied, from which larger sections (contigs) were made, and at the final stage, the structure of individual chromosomes. The nuclear DNA of the Arabidopsis genome is distributed among five chromosomes. In 1999, the results of sequencing two chromosomes were published, and the publication of information about the primary structure of the remaining three completed the sequencing of the entire genome.
Of 125 million nucleotide pairs, the primary structure of 119 million has been determined, which is 92% of the entire genome. Only 8% of the Arabidopsis genome, containing large blocks of repeating DNA sections, turned out to be inaccessible for study. In terms of completeness and thoroughness of sequencing of eukaryotic genomes, Arabidopsis remains in the top three champions along with the unicellular yeast organism Saccharomyces cerevisiae and multicellular animal organism Caenorhabditis elegance(see table).
About 15 thousand individual genes encoding proteins were found in the Arabidopsis genome. Approximately 12 thousand of these are contained in two copies per haploid (single) genome, so the total number of genes is 27 thousand. The number of genes in Arabidopsis is not much different from the number of genes in organisms such as humans and mice, but the size of its genome 25-30 times less. This circumstance is associated with important features in the structure of individual Arabidopsis genes and the overall structure of its genome.
Arabidopsis genes are compact, containing only a few exons (protein-coding regions), separated by short (about 250 bp) non-coding DNA stretches (introns). The gaps between individual genes average 4.6 thousand nucleotide pairs. For comparison, we point out that human genes contain many tens and even hundreds of exons and introns, and intergenic regions have sizes of 10 thousand nucleotide pairs or more. It is believed that the presence of a small compact genome contributed to the evolutionary stability of Arabidopsis, since its DNA became less of a target for various damaging agents, in particular, for the introduction of virus-like repeating DNA fragments (transposons) into the genome.
Other molecular features of the Arabidopsis genome include the enrichment of exons with guanine and cytosine (44% in exons and 32% in introns) compared to animal genes, as well as the presence of twice repeated (duplicated) genes. It is believed that this doubling occurred as a result of four simultaneous events, which consisted in the doubling (repetition) of part of the Arabidopsis genes, or the fusion of related genomes. These events, which took place 100-200 million years ago, are a manifestation of the general tendency towards polyploidization (a multiple increase in the number of genomes in an organism), characteristic of plant genomes. However, some facts show that in Arabidopsis the duplicated genes are nonidentical and function differently, which may be due to mutations in their regulatory regions.
Another object of complete DNA sequencing was rice. The genome of this plant is also small (12 chromosomes, giving a total of 420-470 million bp), only 3.5 times larger than that of Arabidopsis. However, unlike Arabidopsis, rice is of enormous economic importance, being the basis of nutrition for more than half of humanity, therefore not only billions of consumers are vitally interested in improving its properties, but also a multimillion-dollar army of people actively involved in the very labor-intensive process of growing it.
Some researchers began studying the rice genome back in the 80s of the last century, but this work reached a serious scale only in the 90s. In 1991, a program to decipher the structure of the rice genome was created in Japan, combining the efforts of many research groups. In 1997, on the basis of this program, the International Rice Genome Project was organized. Its participants decided to concentrate their efforts on sequencing one of the rice subspecies ( Oriza sativajaponica), in the study of which significant progress had already been achieved by that time. The Human Genome program became a serious incentive and, figuratively speaking, a guiding star for such work.
As part of this program, the strategy of “chromosomal” hierarchical division of the genome, which participants in the international consortium used to decipher the rice genome, was tested. However, if, when studying the human genome, fractions of individual chromosomes were isolated using various techniques, then material specific to individual rice chromosomes and their individual sections was obtained by laser microdissection (cutting out microscopic objects). On the microscope slide where the rice chromosomes are located, under the influence of a laser beam, everything except the chromosome or its sections intended for analysis is burned out. The remaining material is used for cloning and sequencing.
Numerous reports have been published on the results of sequencing individual fragments of the rice genome, carried out with high accuracy and detail characteristic of hierarchical technology. It was believed that determination of the complete primary structure of the rice genome would be completed by the end of 2003-mid-2004 and the results, together with data on the primary structure of the Arabidopsis genome, would be widely used in the comparative genomics of other plants.
However, in early 2002, two research groups - one from China, the other from Switzerland and the United States - published the results of full rough (rough) sequencing of the rice genome, performed using total cloning technology. In contrast to a step-by-step (hierarchical) study, the total approach is based on the simultaneous cloning of the entire genomic DNA in one of the viral or bacterial vectors and obtaining a significant (huge for medium and large genomes) number of individual clones containing different DNA segments. Based on the analysis of these sequenced sections and the overlapping of identical end sections of DNA, a contig is formed - a chain of DNA sequences joined together. The general (total) contig represents the primary structure of the entire genome or, at least, of an individual chromosome.
In such a schematic presentation, the strategy of total cloning seems uncomplicated. In fact, it encounters serious difficulties associated with the need to obtain a huge number of clones (it is generally accepted that the genome or its region being studied must be overlapped by clones at least 10 times), a gigantic volume of sequencing and the extremely complex work of joining clones, which requires the participation of bioinformatics specialists. A serious obstacle to total cloning is the variety of repeating DNA regions, the number of which, as already mentioned, increases sharply as the genome size increases. Therefore, the total sequencing strategy is used primarily in studying the genomes of viruses and microorganisms, although it was successfully applied to study the genome of a multicellular organism, Drosophila.
The results of total sequencing of this genome were “superimposed” on a huge array of information about its chromosomal, gene and molecular structure obtained over an almost 100-year period of studying Drosophila. And yet, in terms of the degree of sequencing, the Drosophila genome (66% of the total genome size) is significantly inferior to the Arabidopsis genome (92%), despite their fairly similar sizes - 180 million and 125 million nucleotide pairs, respectively. Therefore, it has recently been proposed to call the technology used to sequence the Drosophila genome mixed.
To sequence the rice genome, the above-mentioned research groups took two of its subspecies, the most widely cultivated in Asian countries - Oriza saliva L. ssp indicaj And Oriza saliva L. sspjaponica. The results of their research coincide in many ways, but also differ in many ways. Thus, representatives of both groups stated that they achieved contig overlap of approximately 92-93% of the genome. It has been shown that about 42% of the rice genome is represented by short DNA repeats consisting of 20 nucleotide pairs, and the majority of mobile DNA elements (transposons) are located in intergenic regions. However, information about the size of the rice genome varies significantly.
For the Japanese subspecies, the genome size is determined to be 466 million nucleotide pairs, and for the Indian subspecies - 420 million. The reason for this discrepancy is not clear. It may be a consequence of different methodological approaches to determining the size of the non-coding part of genomes, that is, it may not reflect the true state of affairs. But it is possible that a 15% difference in the size of the studied genomes really exists.
The second serious discrepancy was revealed in the number of detected genes: for the Japanese subspecies - from 46,022 to 55,615 genes per genome, and for the Indian subspecies - from 32,000 to 50,000. The reason for this discrepancy is not clear.
The incompleteness and inconsistency of the information received is noted in the comments to the published articles. It is also hoped that the gaps in knowledge of the rice genome will be eliminated by comparing the data from “rough sequencing” with the results of detailed, hierarchical sequencing carried out by participants in the International Rice Genome Project.
COMPARATIVE AND FUNCTIONAL GENOMICS OF PLANTS
The extensive data obtained, half of which (the results of the Chinese group) are publicly available, undoubtedly open up broad prospects both for the study of the rice genome and for plant genomics in general. A comparison of the properties of the Arabidopsis and rice genomes showed that most of the genes (up to 80%) identified in the Arabidopsis genome are also found in the rice genome, however, for approximately half of the genes found in rice, analogues (orthologs) have not yet been found in the Arabidopsis genome . At the same time, 98% of genes whose primary structure has been established for other cereals have been identified in the rice genome.
The significant (almost twofold) discrepancy in the number of genes in rice and Arabidopsis is puzzling. At the same time, the data from the rough transcript of the rice genome, obtained using total sequencing, are practically not compared with the extensive results of studying the rice genome using the method of hierarchical cloning and sequencing, that is, what has been done for the Drosophila genome has not been achieved. Therefore, it remains unclear whether the difference in the number of genes in Arabidopsis and rice reflects the true state of affairs or is explained by differences in methodological approaches.
Unlike the Arabidopsis genome, information about twin genes in the rice genome is not provided. It is possible that their relative abundance may be greater in rice than in Arabidopsis. This possibility is indirectly supported by data on the presence of polyploid forms of rice. Greater clarity on this issue can be expected after the completion of the International Rice Genome Project and obtaining a detailed picture of the primary DNA structure of this genome. Serious grounds for such hope are given by the fact that after the publication of works on the rough sequencing of the rice genome, the number of publications on the structure of this genome sharply increased, in particular, information appeared on the detailed sequencing of its chromosomes 1 and 4.
Knowing, at least approximately, the number of genes in plants is of fundamental importance for comparative plant genomics. At first it was believed that since all flowering plants are very close to each other in their phenotypic characteristics, their genomes should also be close. And if we study the Arabidopsis genome, we will get information about most genomes of other plants. Indirect confirmation of this assumption is provided by the results of sequencing the mouse genome, which is surprisingly close to the human genome (about 30 thousand genes, of which only 1 thousand turned out to be different).
It can be assumed that the reason for the differences in the genomes of Arabidopsis and rice lies in their belonging to different classes of plants - dicotyledons and monocotyledons. To clarify this issue, it is extremely desirable to know at least the rough primary structure of some other monocot plant. The most realistic candidate may be corn, whose genome is approximately equal to the human genome, but still significantly smaller than the genomes of other cereals. The food value of corn is well known.
The enormous material obtained from sequencing the genomes of Arabidopsis and rice is gradually becoming the basis for a large-scale study of plant genomes using comparative genomics methods. Such studies have general biological significance, as they make it possible to establish the main principles of the organization of the plant genome as a whole and their individual chromosomes, to identify common features of the structure of genes and their regulatory regions, and to consider the relationship between the functionally active (gene) part of the chromosome and various non-protein-coding intergenic DNA regions. Comparative genetics is also becoming increasingly important for the development of human functional genomics. It is for comparative studies that the genomes of puffer fish and mice were sequenced.
No less important is the study of individual genes responsible for the synthesis of individual proteins that determine specific functions of the body. It is in the detection, isolation, sequencing and establishment of the function of individual genes that the practical, primarily medical, significance of the Human Genome program lies. This circumstance was noted several years ago by J. Watson, who emphasized that the Human Genome program will be completed only when the functions of all human genes are determined.

Rice. 4. Classification by function of Arabidopsis genes
1 - genes for growth, division and DNA synthesis; 2 - RNA synthesis genes (transcription); 3 - genes for protein synthesis and modification; 4 - genes for development, aging and cell death; 5 - genes of cellular metabolism and energy metabolism; 6 - genes for intercellular interaction and signal transmission; 7 - genes for supporting other cellular processes; 8 - genes with unknown functionWhen it comes to the function of plant genes, we know less than one-tenth of what we know about human genes. Even in Arabidopsis, whose genome is much more studied than the human genome, the function of almost half of its genes remains unknown (Fig. 4). Meanwhile, plants, in addition to genes common to animals, have a significant number of genes specific only (or at least predominantly) to them. We are talking about genes involved in water transport and the synthesis of cell walls, which are absent in animals, about genes that ensure the formation and functioning of chloroplasts, photosynthesis, nitrogen fixation and the synthesis of numerous aromatic products. This list can be continued, but it is already clear how difficult the task is facing plant functional genomics.
Complete genome sequencing provides close to true information about the total number of genes of a given organism, allows more or less detailed and reliable information about their structure to be placed in data banks, and facilitates the work of isolating and studying individual genes. However, genome sequencing does not mean establishing the function of all genes.
One of the most promising approaches of functional genomics is based on identifying working genes on which mRNA transcription (reading) occurs. This approach, including the use of modern microarray technology, makes it possible to simultaneously identify up to tens of thousands of functioning genes. Recently, using this approach, the study of plant genomes has begun. For Arabidopsis, it was possible to obtain about 26 thousand individual transcripts, which greatly facilitates the possibility of determining the function of almost all of its genes. In potatoes, it was possible to identify about 20,000 thousand working genes that are important for understanding both the processes of growth and tuber formation, and the processes of potato disease. It is expected that this knowledge will improve the resistance of one of the most important food products to pathogens.
A logical development of functional genomics is proteomics. This new field of science studies the proteome, which typically refers to the complete set of proteins in a cell at a given time. This set of proteins, reflecting the functional state of the genome, changes all the time, while the genome remains unchanged.
The study of proteins has long been used to make judgments about the activity of plant genomes. As is known, enzymes found in all plants differ in the sequence of amino acids in individual species and varieties. Such enzymes, with the same function, but different sequences of individual amino acids, are called isoenzymes. They have different physicochemical and immunological properties (molecular weight, charge), which can be detected using chromatography or electrophoresis. For many years, these methods have been successfully used to study so-called genetic polymorphism, that is, differences between organisms, varieties, populations, species, in particular wheat and related forms of cereals. However, recently, due to the rapid development of DNA analysis methods, including sequencing, the study of protein polymorphism has been replaced by the study of DNA polymorphism. However, direct study of the spectra of storage proteins (prolamins, gliadins, etc.), which determine the basic nutritional properties of cereals, remains an important and reliable method for genetic analysis, selection and seed production of agricultural plants.
Knowledge of genes, the mechanisms of their expression and regulation is extremely important for the development of biotechnology and the production of transgenic plants. It is known that impressive successes in this area cause mixed reactions from the environmental and medical communities. However, there is an area of plant biotechnology where these fears, if not completely groundless, then, in any case, seem insignificant. We are talking about creating transgenic industrial plants that are not used as food products. India recently harvested its first crop of transgenic cotton that is resistant to a number of diseases. There is information about the introduction of special genes encoding pigment proteins into the cotton genome and the production of cotton fibers that do not require artificial dyeing. Another industrial crop that may be subject to effective genetic engineering is flax. Its use as an alternative to cotton for textile raw materials has been discussed recently. This problem is extremely important for our country, which has lost its own sources of cotton raw materials.
PROSPECTS FOR STUDYING PLANT GENOMES
It is obvious that structural studies of plant genomes will be based on approaches and methods of comparative genomics using the results of deciphering the genomes of Arabidopsis and rice as the main material. A significant role in the development of comparative plant genomics will, without a doubt, be played by the information that sooner or later will be provided by total (rough) sequencing of the genomes of other plants. In this case, comparative plant genomics will be based on establishing genetic relationships between individual loci and chromosomes belonging to different genomes. We will talk not so much about the general genomics of plants, but about the selective genomics of individual chromosomal loci. Thus, it was recently shown that the gene responsible for vernalization is located in the VRn-AI locus of chromosome 5A of hexaploid wheat and the Hd-6 locus of chromosome 3 of rice.
The development of these studies will be a powerful impetus for the identification, isolation and sequencing of many functionally important plant genes, in particular genes responsible for disease resistance, drought resistance, and adaptability to various growing conditions. Functional genomics, based on mass identification (screening) of genes functioning in plants, will be increasingly used.
We can foresee further improvements in chromosomal technologies, primarily the microdissection method. Its use dramatically expands the possibilities of genomic research without requiring huge costs, such as total genome sequencing. The method of localizing individual genes on plant chromosomes using hybridization will become more widespread. in situ. At the moment, its use is limited by the huge number of repeating sequences in the plant genome, and possibly by the peculiarities of the structural organization of plant chromosomes.
In the foreseeable future, chromosomal technologies will also become of great importance for the evolutionary genomics of plants. These technologies, which are relatively inexpensive, make it possible to quickly assess intra- and interspecific variability and study complex allopolyploid genomes of tetraploid and hexaploid wheat and triticale; analyze evolutionary processes at the chromosomal level; investigate the formation of synthetic genomes and the introduction (introgression) of foreign genetic material; identify genetic relationships between individual chromosomes of different species.
The study of plant karyotype using classical cytogenetic methods, enriched by molecular biological analysis and computer technologies, will be used to characterize the genome. This is especially important for studying the stability and variability of the karyotype at the level of not only individual organisms, but also populations, varieties and species. Finally, it is difficult to imagine how one can estimate the number and spectra of chromosomal rearrangements (aberrations, bridges) without the use of differential staining methods. Such studies are extremely promising for monitoring the environment based on the state of the plant genome.
In modern Russia, it is unlikely that direct sequencing of plant genomes will be carried out. Such work, which requires large investments, is unsustainable for our current economy. Meanwhile, information about the structure of the genomes of Arabidopsis and rice, obtained by world science and available in international data banks, is sufficient for the development of domestic plant genomics. It is possible to foresee an expansion of research into plant genomes based on comparative genomics approaches to solve specific problems of breeding and crop production, as well as to study the origin of various plant species of economic importance.
It can be assumed that in domestic breeding practice and plant growing, genomic approaches such as genetic typing (RELF, RAPD, AFLP analyses, etc.), which are quite affordable for our budget, will be widely used. In parallel with direct methods for determining DNA polymorphism, approaches based on the study of protein polymorphism, primarily storage proteins of cereals, will be used to solve problems of genetics and plant breeding. Chromosome technologies will be widely used. They are relatively inexpensive, and their development requires quite moderate investments. In the field of chromosome research, domestic science is not inferior to the world.
It should be emphasized that our science has made a significant contribution to the formation and development of plant genomics [,].
The fundamental role was played by N.I. Vavilov (1887-1943).
In molecular biology and plant genomics, the pioneering contribution of A.N. is obvious. Belozersky (1905-1972).
In the field of chromosome research, it is necessary to note the work of the outstanding geneticist S.G. Navashin (1857-1930), who first discovered satellite chromosomes in plants and proved that it is possible to distinguish individual chromosomes by the characteristics of their morphology.
Another classic of Russian science G.A. Levitsky (1878-1942) described in detail the chromosomes of rye, wheat, barley, peas and sugar beets, introduced the term “karyotype” into science and developed the doctrine of it.
Modern specialists, relying on the achievements of world science, can make a significant contribution to the further development of plant genetics and genomics.
The author expresses his heartfelt gratitude to Academician Yu.P. Altukhov for critical discussion of the article and valuable advice.The work of the team headed by the author of the article was supported by the Russian Foundation for Basic Research (grants No. 99-04-48832; 00-04-49036; 00-04-81086), the Program of the President of the Russian Federation for the support of scientific schools (grants No. 00-115 -97833 and NSh-1794.2003.4) and the Program of the Russian Academy of Sciences "Molecular genetic and chromosomal markers in the development of modern methods of selection and seed production."
LITERATURE
1. Zelenin A.V., Badaeva E.D., Muravenko O.V. Introduction to plant genomics // Molecular biology. 2001. T. 35. pp. 339-348. 2. Pen E. Bonanza for Plant Genomics // Science. 1998. V. 282. P. 652-654. 3. Plant genomics // Proc. Natl. Acad. Sci. USA. 1998. V. 95. P. 1962-2032. 4. Kartel N.A. and etc. Genetics. Encyclopedic Dictionary. Minsk: Technologia, 1999. 5. Badaeva E.D., Friebe B., Gill B.S. 1996. Genome differentiation in Aegilops. 1. Distribution of highly repetitive DNA sequences on chromosomes of diploid species // Genome. 1996. V. 39. P. 293-306. History of chromosome analysis // Biol. membranes. 2001. T. 18. pp. 164-172.
© M.D. Golubovsky
Non-canonical hereditary changes
M.D. Golubovsky
Mikhail Davidovich Golubovsky, Doctor of Biological Sciences, Leading Researcher
St. Petersburg branch of the Institute of History of Natural Science and Technology of the Russian Academy of Sciences.
Genetics as a science took shape 100 years ago, after the secondary discovery of Mendel’s laws. Its rapid development has been marked in recent years by the decoding of the nucleotide composition of the DNA genome of many dozens of species. New branches of knowledge have emerged - genomics, molecular paleogenetics. At the beginning of 2001, as part of an expensive 10-year international program, a fundamental decoding of the human genome was announced. These achievements can perhaps be compared with man's spacewalk and landing on the moon.
Genetic engineering and biotechnology have greatly changed the face of science. Here is an interesting episode, already included in the latest report: “After 1998, an unprecedented race began between the 1,100 scientists of the global Human Genome Project community and the private equity firm Celera Genomics.”. The firm hoped to be the first to reach the finish line and benefit from patenting fragments of human DNA. But so far the principle has won: “What is created by nature and God cannot be patented by man.”
Could Gregor Mendel have imagined such a phantasmagoric picture as he slowly carried out his experiments year after year in the quiet of the monastery garden? To what extent does it transform the natural self-development of science? Does a total DNA analysis of genomes really remove all the covers? Hopes that Pinocchio had already found the treasured golden key to the secret door collided with unforeseen reality and paradoxes. In humans, only 3% of the DNA of the genome encodes proteins, and perhaps another 20-25% is involved in the regulation of gene action. What is the function, and does the rest of the DNA have one? Genes in the genome are sometimes compared to small islands in a sea of inactive and possibly “junk” sequences. The DNA race sometimes resembles the saying: “bring this, I don’t know what.”
The objections of the skeptics are by no means eliminated. Indeed, with total sequencing, the nomination (to use a fashionable term) of a certain DNA segment to “gene rank” is carried out only on the basis of purely formal criteria (genetic punctuation marks necessary for transcription). The role, time and place of action of most “nominee genes” are still completely unclear.
But there is another problem. By genome we must understand the entire hereditary system, including not only the structure of a certain set of DNA elements, but also the nature of the connections between them, which determines the course of ontogenesis in specific environmental conditions. There is a systemic triad: elements, connections between them and properties of integrity. This leads to an important conclusion: knowledge of the structure of genes at the DNA level is necessary, but not at all sufficient to describe the genome. We are only on the threshold of comprehending the dynamic method of organization and non-canonical forms of inheritance [,].
Unexpectedly, at the end of the twentieth century. the question of what the boundaries and spectrum of hereditary variability are has gone beyond purely academic discussions. First in England, and then in Germany, cattle had to be slaughtered due to a neurodegenerative anomaly that could be transmitted to people through the meat of sick animals. The infectious agent turned out to be not DNA or RNA, but proteins called prions (from the English prions - protein infectious particles - protein infectious particles).
Researchers first encountered their unusual manifestation back in the 60s. But then they tried to interpret this phenomenon within the framework of classical ideas, believing that these were “slow viral infections” of animals or a special type of suppressor mutations in yeast. Now it turns out “the prion phenomenon is not an exotic phenomenon characteristic of mammals, but rather a special case of a general biological mechanism” dynamic inheritance. It is likely that the central dogma of molecular genetics will have to be supplemented to take into account the possibility of intra- and interspecies transmission by type of infection.
In the early 80s, the classic of molecular biology and genetics R.B. Khesin identified three forms of non-canonical hereditary variability: non-random ordered changes in loci and chromosome regions consisting of DNA repeats; change and inheritance of cytoplasmic properties; epigenetic inheritance of local and general changes in chromatin packaging. Then mobile genes were added, the behavior of which led to the problem of genome variability.
The purpose of this article is to show that different forms of non-Mendelian inheritance are not an exception, but a consequence of more general ideas about the organization of the genome. Hereditary changes are by no means reduced only to mutations.
Andre Lvov and the role of his discovery
By an amazing coincidence, in the same year of 1953, two articles appeared that determined the face of modern genetics: the discovery of the double helix of DNA by J. Watson and F. Crick and the concept of prophage and lysogeny of bacteria by A. Lvov (1902-1994), which, in my opinion view, is now no less important for biology, medicine and genetics than the double helix of DNA.
Lvov established that the phage can be integrated into the chromosome of a bacterium and transmitted over many generations as an ordinary bacterial gene. In this state, only the repressor gene works in the phage, which blocks the work of all its other loci. A bacterium that has included a phage in its genome is called lysogenic, and the integrated phage is called a prophage. Such a lysogenic bacterium is protected from infection by other phages. Under the influence of ultraviolet radiation or changes in the internal environment of the cell, the repressor is inactivated, the blockade is removed, and the phage multiplies, causing cell death. Now it’s even difficult to imagine how revolutionary this discovery was.
Andre Lvov is a native of Russia; his parents emigrated to France at the end of the 19th century. The image of the scientist’s mother Maria Siminovich is forever captured on the canvas of the artist V. Serov “Girl Illuminated by the Sun” (1888). Maria Yakovlevna Lvova-Siminovich lived to be 90 years old. A few weeks before the Second World War, she donated letters and drawings from V. Serov to the Tretyakov Gallery. Lvov’s father knew Mechnikov and took his son to see him at the Pasteur Institute. This is how the threads of culture stretch and intertwine across centuries and countries. During his long life, A. Lvov worked successively as a protozoologist, bacteriologist, biochemist, geneticist and, finally, as a virologist. At the Pasteur Institute, he patronized both J. Monod and F. Jacob, who shared the 1965 Nobel Prize with the master for the discovery of the operon.
Already since the 20s, strains of bacteria have been known that supposedly carry phages in a latent state and from time to time cause cell lysis. However, the discoverer of bacteriophagy F.D. Herrel looked at the phage only as a cell-lethal agent, not allowing the thought of its latent state. This opinion was initially shared by the classic of molecular genetics M. Delbrück. The fact is that he and his colleagues in the USA worked with so-called T-phages, which are unable to integrate into the chromosome of bacteria. Due to the “demon of authorities,” lysogeny has not been scrupulously studied since the 20s. The pioneer of these works, a brilliant microbiologist from the Pasteur Institute, Eugene Wolman, was captured by the Germans as a Jew during occupation of Paris and died.
After the war, Lvov resumed research into latent phage carriage at the Pasteur Institute. In 1953, he created a coherent concept of the prophage, immediately realizing its significance for the viral theory of cancer and a number of viral pathologies in humans. His clear diagram of the phenomenon of lysogeny is still given in all reports on molecular genetics.
In 1958, F. Jacob and Elias Wolman (son of Eugene Wolman) introduced the term “episome” for elements that can exist either in a free state or integrated into the host genome. They included temperate phages, the sex factor of bacteria, and colicinogenicity factors, with the help of which some strains of bacteria kill other bacteria, as episomes. In the remarkable book “Sex and Genetics of Bacteria,” written in 1961 (and published the following year through the efforts of the famous geneticist S.I. Alikhanyan in Russian translation), the authors foresaw the existence of episome-like elements in higher organisms, shrewdly pointing to “controlling elements”, discovered by B. McClintock in the early 50s (Nobel Prize in Physiology or Medicine 1983). However, at the time they did not realize how deep this analogy was. After the discovery in the early 70s of insertional mutations caused by the inclusion of viral DNA into the cellular genome of bacteria, it became possible to build an evolutionary series of two-way transitions: insertional segments, transposons, plasmids, phages.
Similar series of transformations have been found in eukaryotes. In Drosophila, mobile elements of the gypsy (“gypsy”) family can exist in the form of copies built into the chromosome; be in the form of their full or reduced circular or linear plasmids in the cytoplasm; finally, in the case of individual “permissive” mutations in the host’s genome, they are able to coat themselves with an envelope, become true infectious retroviruses and infect foreign hosts through food. The similarity of P-transposons in Drosophila and the endogenous retrovirus HIV in humans (Table) allows us to predict possible evolutionary genetic events in human populations, the fate of inevitable now and in the future contacts with foreign genomes.
The principle of facultativeness and the generalized concept of the genome
Many facts of variability associated with transposable elements do not fit into the concept of mutations as localized changes in the structure, number or location of gene loci. In order to combine the data of classical and “mobile” genetics, in 1985 I proposed a natural classification of genome elements, including two subsystems: obligate (genes and their regulatory regions in chromosomes) and facultative elements (DNA and RNA carriers, the number and topography of which varies in different cells or organisms of the same species).
Important consequences follow from this classification, making it possible to comprehend or formulate many unusual facts in the field of hereditary variability. Let's name some of them:
- universality of optionality. There are no species genomes that consist only of obligate elements, just as there are no living organisms that consist only of a skeletal skeleton;
- genetic non-identity of daughter cells. Due to randomness, they differ in the number and composition of cytoplasmic facultative elements. The ratio of fractions of obligate and facultative DNA elements is a relatively stable species trait. Having a similar number of gene loci, closely related species can differ in the amount of DNA by 2-5 or more times, increasing blocks of repeats and changing their genomic topography. Various transitions are continuously observed between the obligate and facultative parts of the genome. The most obvious examples are gene mutations due to the introduction (insertions) of transposable elements or multiplication of the number (amplification) of chromosome segments and their transition to different intra- and extrachromosomal states;
- a characteristic type of hereditary variation for each of the two genome subsystems. Morgan mutations are easily correlated with the obligate component. I proposed to call various hereditary changes in the number and topography of optional elements “variations” (as in music - variations on a given theme). Mutations, according to classical concepts, occur, as a rule, randomly, with a low frequency in individual individuals. The nature of the variations is completely different - massive, ordered changes are possible here under the influence of a variety of factors, including weak non-mutagenic factors (temperature, diet, etc.);
- the two-stage nature of most natural hereditary changes. First, facultative elements are activated as the most sensitive to environmental changes. Then gene loci begin to be indirectly affected. We came to this conclusion after many years of observing outbreaks of mutations in nature. Most of them turned out to be unstable and were caused by insertions of transposable elements that are mysteriously activated from time to time in nature. In Drosophila, about 70% of mutations that arise spontaneously in nature or in the laboratory are associated with movements of mobile elements.
The generalized idea of the genome as an ensemble of obligate and facultative elements also expands the concept of “horizontal transfer”, which includes not only the integration of foreign genes into nuclear chromosomes. We can already talk about horizontal transfer in cases of creating a stable association of two genetic systems, in which new characteristics and properties appear.
Functional facultativeness of the genome
Hereditary changes arise as a result of errors in processes that operate with the hereditary material of any living organisms - replication, transcription, translation, as well as repair and recombination.
Facultative replication means the possibility of relatively autonomous hyper- or hypo-replication of individual DNA sections, regardless of the planned regular replication of the entire genomic DNA during cell division. Chromosome sections with repeats, blocks of heterochromatin, have these properties. In this case, autonomous replication leads to an increase in the number of individual segments and, as a rule, has an adaptive nature.
Transcription facultativeness consists in the possibility of the appearance of different mRNAs from the same template due to the presence of more than one promoter and alternative splicing in a given locus. This situation is normal for many genes.
Ambiguity (in the terminology of S.G. Inge-Vechtomov) of translation is manifested in different variants of recognition of the same codon, for example, a stop codon or a codon for including a certain amino acid in the synthesized protein. Such translation depends on the physiological conditions in the cell and on the genotype.
According to the theory of the mutation process by M.E. Lobashev, the occurrence of mutation is associated with the ability of the cell and its hereditary structures to repair damage. It follows that the appearance of a mutation is preceded by a state when the damage is either completely reversible or can be realized in the form of a mutation, understood as “non-identical repair.” By the early 70s, it became clear that the stability of DNA in a cell is not an immanent property of the DNA molecules themselves - it is maintained by a special enzymatic system.
Since the mid-70s, the evolutionary role of “recombination errors” as an inducer of hereditary changes, much more powerful than DNA replication errors, began to become clearer.
At the molecular level, three types of recombination are distinguished: general, site-specific and replicative. For the first, general, regular recombination (crossing over), repair includes breaks in the DNA chain, their cross-linking and restoration. It requires long regions of DNA homology. Site-specific recombination is content with short, several bases, regions of homology, such as, for example, the DNA of phage l and the chromosome of a bacterium. Similarly, the inclusion of mobile elements in the genome and somatic local recombination in ontogenesis between immunoglobulin genes occur, creating their amazing diversity.
Errors in general recombination can be considered as natural consequences of the linearly extended structure of genes. A dilemma arises, which Khesin wrote about: we can assume that mitotic recombinations are a special type of mutagenesis or, on the contrary, some types of mutations (chromosomal aberrations) are the result of “errors” in mitotic recombinations.
If movements of transposable elements or recombination of regions are programmed in ontogenesis, it is difficult to classify such hereditary changes. Sex transformation in yeast has long been considered a mutational event, but it turned out that at a certain stage of ascospore development it occurs with a high probability as a result of site-specific recombination.
Genome variations in response to environmental challenges
In the theory of evolution and in genetics, the question of the connection between hereditary changes and the direction of selection has always been discussed. According to Darwinian and post-Darwinian ideas, hereditary changes occur in different directions and only then are picked up by selection. The replica method, invented in the early 50s by the Lederberg couple, turned out to be especially visual and convincing. Using velvet material, they obtained exact copies - fingerprints - of the experimental inoculation of bacteria on a Petri dish. Then, on one of the plates, selection was carried out for resistance to the phage and the topography of the points of appearance of resistant bacteria on the plate with the phage and in the control was compared. The location of phage-resistant colonies was identical in the two replica dishes. The same result was obtained when analyzing positive mutations in bacteria defective in any metabolite.
Discoveries in the field of mobile genetics have shown that a cell as an integral system can adaptively rearrange its genome during selection. She is able to respond to the challenge of the environment with an active genetic search, and not passively wait for the random occurrence of a mutation that allows her to survive. And in the experiments of the Lederberg spouses, the cells had no choice: either death or adaptive mutation.
In those cases where the selection factor is not lethal, gradual genome rearrangements are possible, directly or indirectly related to the selection conditions. This became clear with the discovery in the late 70s of a gradual increase in the number of loci in which genes for resistance to a selective agent that blocks cell division are located. It is known that methotrexate, an inhibitor of cell division, is widely used in medicine to stop the growth of malignant cells. This cellular poison inactivates the enzyme dihydrofolate reductase (DHFR), the operation of which is controlled by a specific gene.
The resistance of Leishmania cells to the cytostatic poison (methotrexate) increased stepwise, and the proportion of amplified segments with the resistance gene increased proportionally. Not only the gene being selected was multiplied, but also large sections of DNA adjacent to it, called amplicons. When Leishmania venom resistance increased 1000-fold, the amplified extrachromosomal segments accounted for up to 10% of the DNA in the cell! We can say that a pool of facultative elements was formed from one obligate gene. An adaptive restructuring of the genome occurred during selection.
If selection continued long enough, some of the amplicons were integrated into the original chromosome, and after the selection ceased, the increased resistance was persistently maintained.
With the removal of the selective agent from the environment, the number of amplicons with the resistance gene gradually decreased over a number of generations and, at the same time, resistance decreased. Thus, the phenomenon of long-term modifications was modeled, when massive changes caused by the environment are inherited, but gradually fade away over a number of generations.
During repeated selection, some of the amplicons preserved in the cytoplasm ensured their rapid autonomous replication, and resistance arose much faster than at the beginning of the experiments. In other words, a unique cellular amplicon memory of the past selection was formed on the basis of the preserved amplicons.
If we compare the replica method and the course of selection for resistance in the case of amplification, it turns out that it was contact with the selective factor that caused the transformation of the genome, the nature of which correlated with the intensity and direction of selection.
Discussion about adaptive mutations
In 1988, an article by J. Cairns and co-authors appeared in the journal Nature about the occurrence of selection-dependent “directed mutations” in the bacterium E. coli. We took bacteria that carried mutations in the lacZ gene of the lactose operon, unable to break down the disaccharide lactose. But these mutants could divide on a medium with glucose, from where, after one or two days of growth, they were transferred to a selective medium with lactose. Having selected lac+ reverses, which, as expected, arose during the “glucose” divisions, non-growing cells were left under conditions of carbohydrate starvation. At first, the mutants died off. But after a week or more, new growth was observed due to an outbreak of reversions specifically in the lacZ gene. It was as if cells under conditions of severe stress, without dividing (!), were conducting a genetic search and adaptively changing their genome.
B. Hall's subsequent work used bacteria mutant for the tryptophan utilization gene (trp). They were placed on a medium devoid of tryptophan, and the frequency of reversions to normal was assessed, which increased precisely during tryptophan starvation. But the cause of this phenomenon was not the starvation conditions themselves, because on the medium with cysteine starvation, the frequency of reversions to trp+ did not differ from the norm.
In the next series of experiments, Hall took double tryptophan-deficient mutants, carrying both mutations in the trpA and trpB genes, and again placed the bacteria on a medium devoid of tryptophan. Only individuals in which reversions occurred simultaneously in two tryptophan genes could survive. The frequency of occurrence of such individuals was 100 million times higher than expected from a simple probabilistic coincidence of mutations in two genes. Hall preferred to call this phenomenon “adaptive mutations” and subsequently showed that they also occur in yeast, i.e. in eukaryotes.
The publications of Cairns and Hall immediately sparked heated debate. The result of its first round was the presentation of one of the leading researchers in the field of mobile genetics, J. Shapiro. He briefly discussed two main ideas. First, the cell contains biochemical complexes, or “natural genetic engineering” systems, that are capable of reconstructing the genome. The activity of these complexes, like any cellular function, can change dramatically depending on the physiology of the cell. Secondly, the frequency of occurrence of hereditary changes is always assessed not for one cell, but for a cell population in which cells can exchange hereditary information with each other. In addition, intercellular horizontal transfer by viruses or transfer of DNA segments is enhanced under stress conditions. According to Shapiro, these two mechanisms explain the phenomenon of adaptive mutations and return it to the mainstream of conventional molecular genetics. What, in his opinion, are the results of the discussion? “We found a genetic engineer there with an impressive array of intricate molecular tools to reorganize the DNA molecule.” .
Recent decades have revealed an unforeseen realm of complexity and coordination at the cellular level that is more compatible with computer technology than with the mechanized approach that dominated the creation of the neo-Darwinian modern synthesis. Following Shapiro, there are at least four groups of discoveries that have changed the understanding of cellular biological processes.
Genome organization. In eukaryotes, genetic loci are arranged according to a modular principle, representing structures of regulatory and coding modules common to the entire genome. This ensures the rapid assembly of new constructs and the regulation of gene ensembles. Loci are organized into hierarchical networks, led by a major switch gene (as in the case of sex regulation or eye development). Moreover, many of the subordinate genes are integrated into different networks: they function at different periods of development and influence many phenotypic traits.Under conditions of environmental challenge, the cell acts purposefully, like a computer, when when it starts, the normal operation of the main programs is checked step by step, and in the event of a malfunction, the computer’s operation stops. In general, it becomes obvious, already at the cellular level, that the unconventional French evolutionary zoologist Paul Grasset is right: “To live means to react, and not to be a victim.”Reparative capabilities of the cell. Cells are not at all passive victims of random physicochemical influences, since they have a repair system at the level of replication, transcription and translation.
Mobile genetic elements and natural genetic engineering. The work of the immune system is based on the continuous construction of new variants of immunoglobulin molecules based on the action of natural biotechnological systems (enzymes: nucleases, ligases, reverse transcriptases, polymerases, etc.). These same systems use mobile elements to create new inheritable structures. At the same time, genetic changes can be massive and ordered. Genome reorganization is one of the basic biological processes. Natural genetically engineered systems are regulated by feedback systems. For the time being, they remain in an inactive state, but during key periods or during stress they are brought into action.
Cellular information processing. Perhaps one of the most important discoveries in cell biology is that a cell continuously collects and analyzes information about its internal state and external environment to make decisions about growth, movement, and differentiation. Particularly indicative are the mechanisms of control of cell division that underlie growth and development. The process of mitosis is universal in higher organisms and includes three successive stages: preparation for division, chromosome replication and completion of cell division. Analysis of the gene control of these phases led to the discovery of special points at which the cell checks whether the repair of violations in the DNA structure occurred at the previous stage or not. If errors are not corrected, the next stage will not start. When the damage cannot be eliminated, a genetically programmed system of cell death, or apoptosis, is launched.
Ways of occurrence of natural hereditary changes in the environment-facultative elements-obligate elements system. Facultative elements are the first to perceive non-mutagenic environmental factors, and then the variations that arise cause mutations. The behavior of facultative elements is also influenced by obligate elements.
Non-canonical hereditary changes that arise under the influence of selection for cytostatics and lead to gene amplification.
Acquired characteristics are inherited
“The history of biology does not know a more expressive example of a centuries-old discussion of a problem than the discussion about inheritance or non-inheritance of acquired characteristics,”- these words appear at the beginning of the book by the famous cytologist and historian of biology L.Ya. Blyakher. In history, perhaps, we can recall a similar situation with attempts to transform chemical elements. Alchemists believed in this possibility, but in chemistry the postulate about the immutability of chemical elements was established. However, nowadays in nuclear physics and chemistry, research on the transformation of elements and analysis of their evolution is commonplace. Who turned out to be right in a centuries-old dispute? We can say that at the level of chemical molecular interactions there is no transformation of elements, but at the nuclear level it is a rule.
A similar analogy arises with the question of the inheritance of characteristics that appeared during ontogenesis. If newly emerging hereditary changes are reduced only to mutations of genes and chromosomes, then the question can be considered closed. But if we proceed from the generalized concept of the genome, including the idea of dynamic heredity [,], the problem needs to be revised. In addition to mutation, there are variational and epigenetic forms of hereditary variability, associated not with changes in the DNA text, but in the state of the gene. Such effects are reversible and heritable.
It is interesting that the International Yearbook on Genetics, published at the end of 1991, opens with an article by O. Landman “Inheritance of acquired characteristics.” The author summarizes the facts obtained in genetics long ago, showing that “The inheritance of acquired characteristics is quite compatible with the modern concept of molecular genetics.” Landman examines in detail about ten experimental systems in which the inheritance of acquired characteristics has been established. Four different mechanisms can lead to it: changes in the structures of the cell membrane, or cortex, studied by T. Sonneborn in ciliates; DNA modifications, i.e. clonally transmitted changes in the nature of local DNA methylation (this includes the phenomenon of imprinting); epigenetic changes without any DNA modifications; induced loss or acquisition of optional elements.
Landman's article makes us, as it were, witnesses to a critical period of change in a postulate in genetics that seemed as unshakable as a rock. The author calmly, without excitement or new stunning facts, combines old and new data into a system, giving them a clear modern interpretation. A general principle can be formulated: inheritance of acquired characteristics is possible in cases where a certain phenotypic character depends on the number or topography of facultative elements.
I will give two instructive examples on Drosophila: the first is associated with the behavior of the sigma virus, the second - with mobile elements responsible for hybrid sterility of females and supermutability.
The study of the interaction of the sigma virus with the Drosophila genome began more than 60 years ago. First, in 1937, the French geneticist F. Léritier discovered sharp hereditary differences in different lines of flies in the degree of sensitivity to carbon dioxide (CO 2 ). The trait was inherited in a bizarre way: through the cytoplasm, but not only through the maternal line, and sometimes through males. Sensitivity could also be transferred by injecting hemolymph into different types of Drosophila. In these cases, the trait was not transmitted stably, but as a result of selection, inheritance became stable.
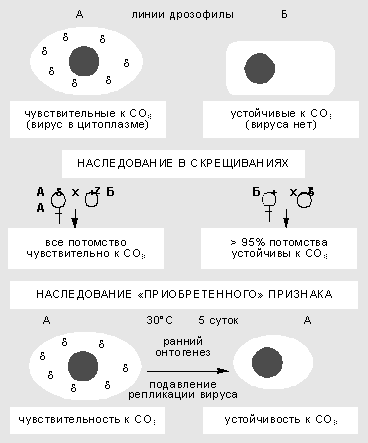
Non-Mendelian inheritance of a trait in Drosophila that depends on a population of facultative genomic elements. The sign of sensitivity to CO 2 is caused by the presence of rhabdovirus sigma in the cytoplasm of the fly. As a result of a temperature shock at an early stage of Drosophila development, the reproduction of the virus is blocked, and the grown individuals acquire resistance to it.Sensitivity to CO 2 was associated with the stable reproduction in germinal and somatic cells of the RNA-containing bullet-shaped rhabdovirus sigma, which is similar in a number of properties to the rabies virus in mammals. Oogonia (cells from which eggs are formed during meiosis and maturation) in females of a stabilized line usually contain 10-40 viral particles, and oocytes (mature eggs) - 1-10 million. The sigma virus is a typical facultative element. Mutations in its genome lead to complex forms of system behavior. Cases of virus carriage have been found in which Drosophila remain resistant to CO 2, but at the same time are immune to infection by other strains of the virus. The situation is quite comparable to the behavior of the phage-bacterium system, which was immediately noticed by F. Jacob and E. Wolman.
The relationship between the Drosophila genome and the virus reproducing in its cytoplasm obeys the rules of intracellular genetics. Impacts during ontogenesis can cause a shift in the number and intercellular topography of particles and, as a result, change the degree of sensitivity to carbon dioxide. Thus, elevated temperature blocks the replication of viral particles. If females and males are kept at a temperature of 30°C for several days during gametogenesis, the offspring of such flies will be free of the virus and resistant to CO 2. This means that a trait acquired during individual development is inherited over a number of generations.
The situation with the sigma virus is not isolated. French geneticists studied the factors of female sterility associated with the behavior of type “I” mobile elements. The inheritance of this trait is determined by complex nuclear-cytoplasmic interactions. If active I-elements are localized in paternal chromosomes, then against the background of R-cytoplasm they begin to be activated, undergo multiple transpositions and, as a result, cause severe ontogenetic disturbances in the offspring of females with sensitive cytoplasm. Such females lay eggs, but some of the embryos die at the early stage of cleavage - even before the formation of the blastomere. Lines isolated from natural populations differ in the strength of the action of I-factors and the degree of reactivity (or sensitivity) of the cytoplasm. These indicators can be changed by external influence. The age of the original parental females, as well as exposure to increased temperature in the early period of development, affects not only the fertility of grown females, but also the fertility of their offspring. Changes in cytoplasmic reactivity caused by environmental conditions are maintained over many cell generations. “The most remarkable thing is that these changes in the reactivity of the cytoplasm under the influence of non-genetic factors are inherited: inheritance of “acquired” characteristics is observed,”- noted R.B. Khesin.
Inheritance through the cytoplasm: from grandmothers to grandchildren
In the theory of development and phenogenetics of the twentieth century. An important place is occupied by the deep and completely original research of embryologist P.G. Svetlov (1892-1972). Let us dwell on the theory of the quantization of ontogenesis developed by him (the presence of critical periods in development, when the determination of morphogenetic processes occurs and at the same time the sensitivity of cells to damaging agents increases) and on the idea developed in connection with this that the study of ontogenesis should not be carried out from the moment of fertilization and the formation of the zygote, and also from gametogenesis, including oogenesis in females of the previous generation - the proembryonic period.
Based on these postulates, Svetlov conducted simple and clear experiments on Drosophila and mice in the 60s. He convincingly showed that persistent non-Mendelian inheritance of cytoplasmic properties is possible, and modifications in the expression of mutant traits that arose after a short-term external influence during a critical period of organism development are also transmitted over a number of generations.
In one of the series of experiments, he compared the degree of manifestation of the mutant trait in the offspring of two lines of mice heterozygous for the recessive mutation of microphthalmia (reduced size of the retina and eyes from the moment of birth): heterozygotes normal in phenotype, whose mothers were mutant, and those with mutant fathers. The offspring from the mutant grandmother were distinguished by a stronger manifestation of the trait. Svetlov explained this strange fact by the fact that the female gametes of heterozygous females were still in the body of their mutant mothers and were influenced by them, which increased the mutations in their grandchildren.
Essentially, Svetlov established a phenomenon that later became known as “genomic imprinting” - the difference in the expression of a gene depending on whether it came to the offspring from the mother or from the father. These works, alas, remained underestimated.
It is interesting that back in the late 80s, imprinting, as K. Sapienza, a researcher of this phenomenon, wittily noted, was “It is generally considered a genetic curiosity, affecting only a very few traits. I have been asked more than once why I simply waste my time on such an insignificant phenomenon.”. Most researchers unconditionally accepted one of Mendel's main provisions - the “germ”, or gene, cannot change its potency depending on gender, on which the commonly observed 3:1 split is based. But Sapienza quite rightly noted that when analyzing Mendelian segregation, they usually consider only the presence or absence of a trait, and if it is quantitative, then the boundary yes or no set according to the accepted threshold. If we determine the degree of manifestation of the trait, the influence of genomic imprinting will be revealed.
This was precisely Svetlov’s approach when he carefully studied how the expression of traits in offspring changes depending on the maternal genotype. As an embryologist, he saw the commonality of hereditary and special non-hereditary changes - phenocopies (simulating mutations), if the same morphogenetic apparatus responsible for the implementation of a given trait is affected.
For the first time, using different animal species (drosophila and mice), Svetlov showed the possibility of inheritance through meiosis of the altered nature of the manifestation of a mutant gene. It is not for nothing that Khesin called these works wonderful in his summary.
Short-term (20 min) heating of the body of an eight-day-old female mouse caused persistent changes in oocytes, weakening the effect of the harmful mutation in grandchildren! “The transfer of improvement in eye development observed in experiments with heating can only be explained by the transfer of properties acquired by inheritance in the oocytes of heated females.”. Svetlov associated this phenomenon with the peculiarities of the formation and structure of the egg in animals, because “in the oocyte there is, as it were, a frame that reflects the most general features of the architectonics of the organism under construction.” To prevent developmental disorders in humans, he substantiated the need to study the critical periods of gametogenesis, in which sensitivity to damage is increased. Perhaps, in the pathogenesis of developmental abnormalities in humans, the stage of gamete formation is even more important than embryogenesis.

Scheme of experiments by P.G. Svetlov demonstrating the transmission of the mutation microphthalmia in a number of generations of mice. A single 20-minute exposure to elevated temperature on eight-day-old mutant mice leads to improved eye development in their offspring (F1 and F2). This trait is inherited only through the maternal line and is associated with changes in oocytes.Today, this conclusion is confirmed by molecular genetic studies of the last decade. Drosophila has three systems of maternal genes that form axial and polar heterogeneity of the cytoplasm and gradients in the distribution of biologically active gene products. Long before the onset of fertilization, molecular determination (predestination) of the structural plan and initial stages of development occurs. The gene products of the cells of the mother's body play an important role in the formation of the oocyte. In some ways, this can be compared to a group of worker bees feeding a queen in a hive.
In humans, primary germ cells, from which egg-gametes then arise, begin to separate in a two-month embryo. At the age of 2.5 months they enter meiosis, but immediately after birth this division is blocked. It resumes after 14-15 years with the onset of puberty, when eggs are released from the follicles once a month. But at the end of the second division, meiosis stops again and its blockage is removed only when it meets sperm. Thus, female meiosis begins at 2.5 months and ends only 20-30 or more years later, immediately after fertilization.
The zygote at the two to eight cell stage has weakened genomic immunity. When studying unstable insertional mutations in natural populations of Drosophila, we found that activation of a transposable element, accompanied by a mutational transition, often occurs already in the first divisions of the zygote or in the first divisions of primordial germ cells. As a result, one mutant event immediately captures a clone of primary germ cells, the pool of gametes becomes mosaic, and hereditary changes in the offspring occur in bundles or clusters, simulating family inheritance.
These experiments are very important for epidemiology, when the question arises about the degree of influence of a particular viral epidemic on the gene pool of the offspring. The pioneering research of S.M. Gershenzon and Yu.N. Aleksandrov, begun in the early 60s, led to the conclusion that DNA and RNA viruses and their nucleic acids are powerful mutagenic agents. When they enter a cell, they provoke genomic stress, activate the host's mobile element system, and cause unstable insertional mutations in a group of selected loci specific to each agent.
Now imagine that we want to assess the impact of a viral pandemic (for example, influenza) on human hereditary variability. At the same time, it can be expected that the frequency of various types of developmental anomalies will be increased in the first generation in offspring born in the year or a year after the epidemic. An assessment of the frequency of mutation and variation changes in germ cells (gametes) should be carried out in the grandchild generation.

Scheme of oogenesis in three successive female generations. P - grandmother, F1 - mother, F2 - daughter.
The overall conclusion is that genetic variation in grandchildren may be highly dependent on the conditions under which oogenesis occurred in their grandmothers! Let's imagine a woman who was about 25 years old in 2000, and who will become a mother in the third millennium. The fertilized egg from which she herself was born began to form while her mother was still a two-month embryo, i.e. somewhere in the mid-50s of the twentieth century. And if the flu was rampant during these years, its consequences should be felt within a generation. To assess the consequences of a global epidemic on the gene pool of humanity, it is necessary to compare the grandchildren of three groups, or cohorts - those whose grandmothers were pregnant in the year when the epidemic broke out, with those whose grandmothers became pregnant before and after the pandemic (these are two control cohorts). Unfortunately, such epidemiological and genetic information important for health protection is not yet available.
About ghosts and fighting monsters
30 years have passed since Svetlov’s experiments, which were simple in technique, but original in concept and profound in their conclusions. In the mid-90s, a psychological turning point occurred: the number of works in the field of hereditary variability, the title of which contains the word “epigenetic,” increased sharply.
Various types of epimutations (hereditary variations in the nature of gene activity that are not associated with changes in the DNA text and are massive, directed and reversible) have moved from the category of marginal to an actively studied phenomenon. It has become obvious that living systems have operational “memory”, which is in continuous contact with the environment and uses the means of natural embryogenetic engineering for a rapid inherited transition from one mode of functioning to another. Living systems are not passive victims of natural selection, and all evolutionary forms of life are not “a blot for a short day of disappearance”, as Mandelstam wrote in his famous masterpiece “Lamarck”.
It turned out that epimutations can often be found in ordinary “classical genes”; you just need to choose a suitable experimental system. Back in 1906, five years before Morgan began working with Drosophila, the French evolutionary biologist L. Cano discovered the Mendelian mutation “corpus luteum” in mice. It had an amazing feature - dominance over normal color (gray-brown) and lethality in homozygote. When heterozygous yellow mice were crossed with each other, due to the death of the homozygotes, normal mice appeared in the offspring in a ratio of not 3:1, but 2:1. Subsequently, it turned out that many dominant mutations behave this way in different organisms.
It turned out that in the transcription region of one of the alleles of the “corpus luteum” gene, a mobile element was introduced that resembles a retrovirus in structure and properties. As a result of this insertion, the gene began to obey the punctuation marks of its intruder and become unpredictably activated “at the wrong time and in the wrong place.” Mutants with insertions develop multiple defects (yellow fur coloration, obesity, diabetes, etc.), and behavior becomes unstable. Unnecessary insertion activity is dampened to varying degrees in different tissues by reversible modification or methylation of DNA bases. At the phenotypic level, the manifestation of the dominant allele varies greatly and is mosaic in nature. Australian geneticists discovered that yellow females selected from a homogeneous line had more yellow mice in their offspring, and the phenotype of the father, the carrier of the mutation, does not affect the color change in the offspring. Females turned out to be more inert, and they, selected for the DNA modification phenotype, or imprints, were better preserved in oogenesis. Other geneticists have also found a purely maternal influence similar to that found in Svetlov’s experiments. Depending on the diet of pregnant females, the severity of the “corpus luteum” mutation changed in a certain way in the genotype of heterozygotes. This altered state is unstable, but is inherited in the offspring. The degree of manifestation of the trait correlated with the degree of methylation of DNA bases in the insert.
Referring to these and other similar experiments, a scientific columnist for Science magazine titled his article “Was Lamarck Still a Little Right?” Such tact is understandable. First, caution is warranted when it comes to revising something that has been considered firmly established for decades. Secondly, the inheritance of acquired characteristics is associated not only with the name of Lamarck, but also with the ghost of Lysenko (the latter is mentioned by the author of the note). Indeed, willingly or unwillingly, the shadow of “Michurin biology” emerges when the problem of inheritance of acquired characteristics is discussed. And not only in Russia, where the memory of the tragedy in biology associated with Lysenko’s dominance is still so vivid.
Today, many generally accepted provisions of classical genetics, which Lysenko rejected, have involuntarily, in defiance of him, become considered almost absolute truth. And yet, if this or that serious researcher discovered something outwardly in tune with Lysenko’s views, he was afraid to make it public, fearing ostracism from the scientific community. And even if the work was published, it was accompanied by many reservations and remained on the periphery of science.
Having become acquainted with the articles of A.A. Lyubishchev (Svetlov’s closest friend) in the 60s, I tried to understand why, being one of the most active samizdat critics of Lysenkoism from 1953 to 1965, his articles and letters were collected in a book “In Defense of Science” (L., 1990) - nevertheless, he did not consider the issue of inheritance of acquired characteristics to be finally resolved. This universally recognized expert in evolutionary biology pointed out the incompleteness of the theory of heredity and the similarity of hereditary and modificational variability. Now we know how difficult it is in many cases to draw a line between them. Lyubishchev cited facts of massive, rapid and orderly transformations of the phenotype in evolution, clearly inexplicable from the standpoint of Morgan's mutations and Darwinian selection. Raising his voice against Lysenko’s monopoly, Lyubishchev spoke out in defense of science as such, against the Arakcheev regime that had established itself in it. In the field of science itself, he followed the ancient principle: "Plato is my friend but the truth is dearer".
9. McClintock B.// Science. 1984. V.226. P.792-801.
10. Cairns J.//Nature. 1988. V.27. P.1-6.
11. Hall D.// Genetics. 1990. V.126. P.5-16
12. Shapiro J.// Science. 1995. V.268. P.373-374.
12. Blyakher L. Ya. The problem of inheritance of acquired characteristics. M., 1971.
13. Landman O.//Ann. Rev. Genet. 1991. V.25. P.1-20.
14. Sokolova K.B. Development of phenogenetics in the first half of the twentieth century. M., 1998.
15. Sapienza K.// In the world of science. 1990. ?12. P.14-20.
16. Svetlov P. G.// Genetics. 1966. ?5. P.66-82.
17. Korochkin L. I. Introduction to developmental genetics. M., 1999.
It is a pandemic parasite that infects 70% of invertebrates worldwide and evolves with them. Most often, the parasite infects insects, where it penetrates their eggs and sperm and is transmitted to their offspring. This fact has led scientists to assume that any genetic changes that arise are passed on from generation to generation.
This discovery, made by scientists led by Jack Werren, indicates that horizontal (interspecies) gene transfer between bacteria and multicellular organisms occurs more often than generally believed, and leaves a certain imprint on the process of evolution. Bacterial DNA can be a full part of an organism's genome and even be responsible for the formation of certain characteristics - at least in invertebrates.
The likelihood that such a large DNA fragment is completely neutral is minimal, and experts believe that the genes it contains provide insects with certain selection advantages. The authors are currently working to identify these benefits. Evolutionary biologists should pay close attention to this discovery.
On 05.09.2011 at 09:36, Limarev said:
Limarev V.N.
Decoding the human genome.
Fragment from the book by L.G. Puchko: “Radiethetic cognition of man”
To solve the problem of deciphering the genome, an international project “human genome” was organized with a budget of billions of dollars.
By 2000, the human genome was virtually mapped. Genes were counted, identified and recorded in databases. These are huge amounts of information.
Recording the human genome in digitized form takes about 300 terabytes of computer memory, which is equivalent to 3 thousand hard drives with a capacity of 100 gigabytes.
It turned out. That a person does not have hundreds of thousands, as previously thought, but just over 30 thousand genes. The fly has fruit flies, there are only half as many of them - about 13 thousand, and the mouse has almost the same number as a person. There are only about 1% of genes unique to humans in the deciphered genome. Most of the DNA helix, as it turned out, is occupied not by genes, but by so-called “empty sections”, in which genes are simply not encoded, as well as double fragments repeated one after another, the meaning and meaning of which is unclear.
In a word, genes turned out to be not even the building blocks of life, but only elements of the blueprint according to which the building of the body is built. Building blocks, as was generally believed before the rise of genetics, are proteins.
It has become absolutely obvious that 1% of genes unique to humans cannot encode such a huge amount of information that distinguishes a person from a mouse. Where is all the information stored? For many scientists, the fact becomes undeniable that without the Divine principle it is impossible to explain human nature. A number of scientists suggest that, within the framework of existing ideas about the human body, it is in principle impossible to decipher the human genome.
The world is not known - it is knowable (my comments on the article).
1) Consider the fragment: “Without the Divine principle, it is impossible to explain human nature.”
The information presented above does not in any way indicate this.
The genome indeed has a more complex structure than previously thought.
But, after all, the computer mentioned in the article does not consist only of memory cells.
A computer has two memories: long-term and operational, as well as a processor in which information is processed. The electromagnetic field is also involved in information processing. In order to decipher genome information, it is necessary to understand how it occurs, not only the storage of information, but also its processing. I also admit the idea that some of the information is stored recorded through an electromagnetic field. And also outside a person, as I already wrote, in special information centers of the Supreme Mind.
Just imagine a continuous text encoded in binary code 0 or 1 in Morse code, while you do not know what language it is written in (English or French....) and you do not know that this continuous text consists of words, sentences, paragraphs, chapters, volumes, shelves, cabinets, etc.
It’s almost the same in biology, only everything here is encoded with a four-digit code and we have so far deciphered the order of elementary genes + - / *, but we don’t know the language and accordingly words, sentences, paragraphs, chapters, volumes, shelves, cabinets, etc. For us, the deciphered genome is still a solid text of 4-grade code and it is almost impossible to study it all head-on.
But it turns out that at certain periods of time (both in the individual and his cohort of generations and in the species, genus) some genes and their complexes (responsible for words, sentences, paragraphs, chapters, volumes, shelves, cabinets, etc.) are active , and in other periods of evolution they are passive, which I indirectly determined by various polygenic characteristics (as shown in the topic General Periodic Law of Evolution).
There are currently only two methods for studying genes, this is a simple laboratory calculation of the sum of genes (DNA) in a sample and there is a device that counts the amount of protein RNA produced stuck to the electronic chip produced specific DNA, but since at any given time a huge amount of DNA is active and, accordingly, a huge number of different proteins are produced through RNA, it is very difficult to separate “these noodles with a spoon, fork and Japanese chopsticks” in this soup and find what you are looking for - find cause-and-effect relationships between specific DNA (as a DNA complex) and its influence on a polygenic trait.
It seems that I have found a simple method of how to sort out this whole soup of DNA, RNA and their proteins that determine the degree of a polygenic trait.
As it turned out, each polygenic trait in the order of evolution of an individual (cohort of generations, species and genus) is periodic; therefore, it must be periodic in the activity of RNA and DNA and therefore you just need to find (first going into genetic details) the correlation between the metric change in the polygenic trait (in individual, cohort of generations, species, genus...) and the corresponding activity of RNA, DNA, proportional to these periods.
Jumping genes
In the middle of the last century, American researcher Barbara McClintock discovered amazing genes in corn that can independently change their position on the chromosomes. Now they are called “jumping genes” or transposable (mobile) elements. The discovery was not recognized for a long time, considering mobile elements to be a unique phenomenon characteristic only of corn. However, it was for this discovery that McClintock was awarded the Nobel Prize in 1983 - today jumping genes have been found in almost all studied species of animals and plants.
Where did jumping genes come from, what do they do in a cell, are they useful? Why, with genetically healthy parents, can a Drosophila fruit fly family, due to jumping genes, produce mutant offspring with high frequency or even be childless? What is the role of jumping genes in evolution?
It must be said that the genes that ensure the functioning of cells are located on chromosomes in a certain order. Thanks to this, it was possible to construct so-called genetic maps for many species of unicellular and multicellular organisms. However, there is an order of magnitude more genetic material between genes than within them! What role this “ballast” part of DNA plays has not been fully established, but it is here that mobile elements are most often found, which not only move themselves, but can also take neighboring DNA fragments with them.

Where do jumping genes come from? It is assumed that at least some of them originate from viruses, since some mobile elements are capable of forming viral particles (for example, the mobile element gipsy in the fruit fly Drosophila melanogaster). Some mobile elements appear in the genome through the so-called horizontal transfer from other species. For example, it has been established that mobile hobo-element (translated into Russian it is called a tramp) Drosophila melanogaster repeatedly reintroduced into the genome of this species. There is a version that some regulatory sections of DNA may also have autonomy and a tendency to “vagrancy”.
Useful ballast
On the other hand, most of the jumping genes, despite the name, behave quietly, although they make up a fifth of the total genetic material Drosophila melanogaster or almost half of the human genome.
The redundancy of DNA, which was mentioned above, has its advantage: ballast DNA (including passive mobile elements) takes the hit if foreign DNA is introduced into the genome. The likelihood that a new element will be integrated into a useful gene and thereby disrupt its function is reduced if there is much more ballast DNA than significant DNA.
Some redundancy of DNA is useful in the same way as the “redundancy” of letters in words: we write “Maria Ivanovna”, but say “Marivan”. Some letters are inevitably lost, but the meaning remains. The same principle works at the level of significance of individual amino acids in a protein-enzyme molecule: only the sequence of amino acids that forms the active center is strictly conserved. Thus, at different levels, redundancy turns out to be a kind of buffer that provides a reserve of system strength. This is how mobile elements that have lost mobility turn out to be not useless for the genome. As they say, “from a thin sheep at least a tuft of wool,” although perhaps another proverb would be better suited here - “every bast in a line.”

Mobile elements that have retained the ability to jump move along Drosophila chromosomes with a frequency of 10–2–10–5 per gene per generation, depending on the type of element, genetic background and external conditions. This means that one out of a hundred jumping genes in a cell can change its position after the next cell division. As a result, after several generations, the distribution of mobile elements along the chromosome can change very significantly.
It is convenient to study this distribution on polytene (multi-stranded) chromosomes from the salivary glands of Drosophila larvae. These chromosomes are many times thicker than usual, which greatly simplifies their examination under a microscope. How are such chromosomes obtained? In the cells of the salivary glands, the DNA of each chromosome is multiplied, as during normal cell division, but the cell itself does not divide. As a result, the number of cells in the gland does not change, but over 10-11 cycles, several thousand identical DNA strands accumulate in each chromosome.

It is partly due to polytene chromosomes that jumping genes in Drosophila are better studied than in other multicellular organisms. As a result of these studies, it turned out that even within the same Drosophila population it is difficult to find two individuals that have chromosomes with the same distribution of transposable elements. It is no coincidence that it is believed that most of the spontaneous mutations in Drosophila are caused by the movement of these “jumpers”.
The consequences may vary...
Based on their effect on the genome, active mobile elements can be divided into several groups. Some of them perform functions that are extremely important and useful for the genome. For example, telomeric The DNA located at the ends of chromosomes in Drosophila consists of special mobile elements. This DNA is extremely important - its loss entails the loss of the entire chromosome during cell division, which leads to cell death.
Other mobile elements are outright “pests”. At least that is what they are considered to be at the moment. For example, mobile elements of the R2 class can be specifically incorporated into arthropod genes encoding one of the ribosomal proteins, the cellular “factories” for protein synthesis. Individuals with such disorders survive only because only a portion of the many genes encoding these proteins are damaged in the genome.

There are also mobile elements that move only in reproductive tissues that produce germ cells. This is explained by the fact that in different tissues the same mobile element can produce enzyme protein molecules required for movement that differ in length and function.
An example of the latter is the P-element Drosophila melanogaster, which entered its natural populations through horizontal transfer from another species of Drosophila no more than a hundred years ago. However, there is hardly a population on Earth now Drosophila melanogaster, in which the P-element would not be found. It should be noted that most of its copies are defective, moreover, the same version of the defect was found almost everywhere. The role of the latter in the genome is unique: it is “intolerant” towards its fellows and plays the role of a repressor, blocking their movement. So the protection of the Drosophila genome from the jumps of the “stranger” may be partially carried out by its own derivatives.
The main thing is to choose the right parents!
Most of the jumps of mobile elements do not affect the appearance of the Drosophila, because they occur on ballast DNA, but there are other situations when their activity increases sharply.

Surprisingly, the most powerful factor inducing the movement of jumping genes is poor parental selection. For example, what happens if you cross females from a laboratory population? Drosophila melanogaster, which do not have the P-element (because their ancestors were caught from nature about a hundred years ago), with males carrying the P-element? In hybrids, due to the rapid movement of the mobile element, a large number of different genetic disorders can appear. This phenomenon, called hybrid dysgenesis, is caused by the fact that there is no repressor in the maternal cytoplasm that prohibits the movement of the transposable element.
Thus, if grooms from population A and brides from population B can create large families, then the opposite is not always true. A family of genetically healthy parents can produce a large number of mutant or infertile offspring, or even be childless if the father and mother have a different set of mobile elements in their genome. Especially many violations appear if the experiment is carried out at a temperature of 29° C. The influence of external factors, superimposed on the genetic background, enhances the effect of genome mismatch, although these factors themselves (even ionizing radiation) alone are not capable of causing such massive movements of mobile elements.
Similar events in Drosophila melanogaster may occur with the participation of other families of mobile elements.
"Mobile" evolution
The cellular genome can be considered as a kind of ecosystem of permanent and temporary members, where neighbors not only coexist, but also interact with each other. The interaction of host genes with mobile elements is still poorly understood, but many results can be given - from the death of the organism in the event of damage to an important gene to the restoration of previously damaged functions.
It happens that the jumping genes themselves interact with each other. Thus, a phenomenon resembling immunity is known, when a mobile element cannot penetrate in close proximity to an already existing one. However, not all mobile elements are so delicate: for example, P-elements can easily penetrate each other and take their fellow players out of the game.

In addition, there is a kind of self-regulation in the number of mobile elements in the genome. The fact is that mobile elements can exchange homologous regions with each other - this process is called recombination. As a result of such interaction, mobile elements may, depending on their orientation, lose ( deletion) or expand ( inversion) fragments of host DNA located between them. If a significant piece of a chromosome is lost, the genome will die. In the case of an inversion or small deletion, chromosome diversity is created, which is considered a necessary condition for evolution.
If recombinations occur between mobile elements located on different chromosomes, the result is the formation of chromosomal rearrangements, which during subsequent cell divisions can lead to genome imbalance. And an unbalanced genome, like an unbalanced budget, is divided very poorly. So the death of unsuccessful genomes is one of the reasons why active mobile elements do not fill chromosomes indefinitely.
A natural question arises: how significant is the contribution of mobile elements to evolution? Firstly, most of the mobile elements are introduced, roughly speaking, wherever they need to be, as a result of which they can damage or change the structure or regulation of the gene into which they are introduced. Then natural selection rejects unsuccessful options, and successful options with adaptive properties are fixed.

If the consequences of the introduction of a mobile element turn out to be neutral, then this variant can persist in the population, providing some diversity in the gene structure. This can come in handy in unfavorable conditions. Theoretically, with the massive movement of mobile elements, mutations can appear in many genes simultaneously, which can be very useful in case of a sharp change in living conditions.
So, to summarize: there are many mobile elements in the genome and they are different; they can interact both with each other and with host genes; can harm and be irreplaceable. Genome instability caused by the movement of mobile elements can end in tragedy for the individual, but the ability to change quickly is a necessary condition for the survival of a population or species. Thanks to this, diversity is created, which is the basis for natural selection and subsequent evolutionary transformations.
An analogy can be drawn between jumping genes and immigrants: some immigrants or their descendants become equal citizens, others are given residence permits, and others - those who do not comply with the laws - are deported or imprisoned. And mass migrations of people can quickly change the state itself.
Literature
Ratner V. A., Vasilyeva L. A. Induction of transpositions of mobile genetic elements by stress influences. Russian binding. 2000.
Gvozdev V. A. Mobile DNA of eukaryotes // Soros educational journal. 1998. No. 8.